
On 11 January 2022, the EU’s Health Technology Assessment Regulation (2021/2282) came into effect following years of negotiating European rules for health technology assessment (HTA). The EU hopes that the new rules will give patients broad, timely access to vital and innovative health technologies, particularly in light of the COVID-19 pandemic. It also believes this will lead to efficiency gains for both health technology developers and Member States – largely through joint clinical assessments coordinated by a body known as the Coordination Group that are to be carried out for medical devices and in vitro diagnostic medical decides from January 2025 at the earliest. Despite that date still being three years away, it is already worth taking a look at the key elements of the HTA Regulation and its implementation in German medical device and social insurance law.
I. Assessing health technologies
Simply put, health technology assessment is a process for systematically and transparently collecting, classifying, analysing and assessing information about a specific health technology, the way it works, and its impact on a healthcare system. The term “health technology” is used very broadly to cover not only medicines, medical devices and in vitro diagnostic medical devices, but also surgical procedures, disease prevention, diagnosis and treatment. HTAs typically examine:
- medical or clinical aspects (in particular the relative effectiveness and relative safety compared to other technologies),
- health economic aspects (in particular the costs and savings compared to other technologies),
- legal aspects (e.g. approval; liability; patient autonomy; data protection),
- social aspects (e.g. access barriers for certain groups of people),
- ethical aspects (e.g. stigmatisation issues; prioritisation of resources)
- and/or organisational aspects (e.g. integration of the technology with existing structures; training of healthcare staff). With the exception of the medical aspects, these areas are also referred to as “non-clinical assessment”.
The resulting HTA report is used as a basis for developing healthcare policies and assisting healthcare decision-makers in choosing whether to provide the health technology as a part of standard care and on what terms.
II. New, joint framework for HTA process in the EU
To date, these HTA processes have been commissioned and conducted at national level – in the case of Germany, by the Federal Joint Committee (G-BA) and the Institute for Quality and Efficiency in Health Care (IQWiG). However, the individual processes themselves and the requirements for scientific evidence that health technology developers need to submit for the HTA has been able to vary greatly between Member States. The industry has therefore faced the difficulty of designing studies – for medicinal products, starting with marketing authorisation – that promise to deliver the best results.
Regulation (EU) 2021/2282 establishes a new, EU-funded framework upon which to operate an HTA process at European level and avoid duplicate effort by health technology developers and Member States, providing a formal upgrade to the voluntary, project-based cooperation of past decades. Key elements of the HTA Regulation include
- the establishment of a coordination group and a stakeholder network,
- conducting joint clinical assessments and joint scientific consultations,
- and supporting voluntary cooperation beyond those elements.
1. Establishment of a coordination group (Article 3)
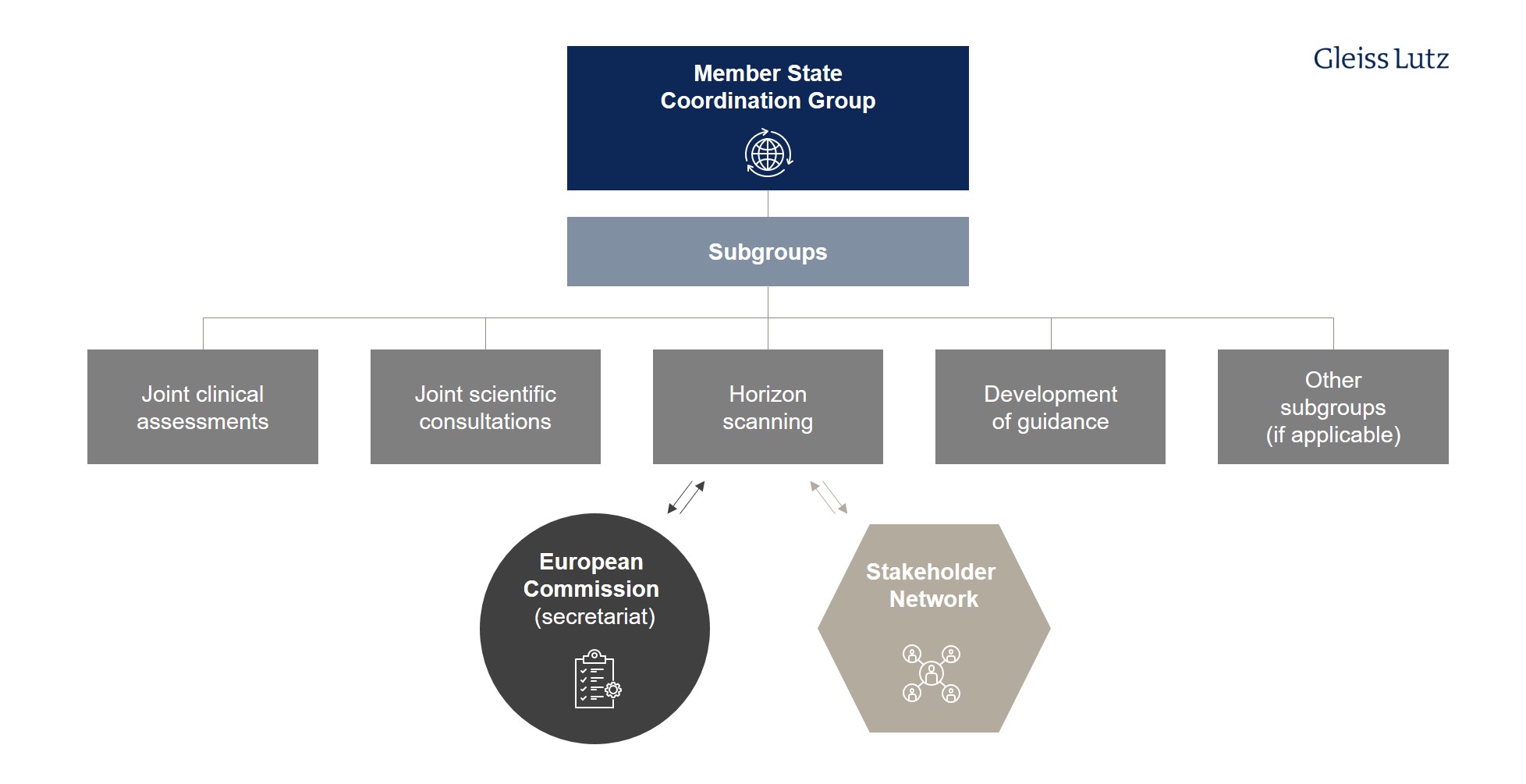
Alongside the Commission, the central actor of the HTA Regulation is the body known as the Coordination Group, which is defined in Article 3. Its members – generally national HTA bodies akin to the Federal Joint Committee and Institute for Quality and Efficiency in Health Care – are designated by the Member States.
- The tasks of the Coordination Group and its subgroups include joint clinical assessment and joint scientific consultations, identifying emerging health technologies (known as horizon scanning), and supporting voluntary cooperation between Member States.
- The Coordination Group is also responsible for developing and adopting the methodological guidance, procedural steps, and timeframes on the basis of which the joint assessments and consultations are performed (Article 3(7), letter (d)-(g)).
- The Regulation itself lays down only rudimentary requirements in this respect. The Coordination Group has a transitional period of three years for this step; under Article 36(2), the Regulation will not apply until 12 January 2025.
The Coordination Group will be supported in its activities by the Commission, which will act as its secretariat. The first meetings are scheduled to take place in June 2022. Here you can find a graphical overview of the different tasks and subgroups of the HTA Coordination Group at EU level.
2. Joint clinical assessment (Articles 7-15)
At the heart of the HTA Regulation is the joint clinical assessment (JCA) of certain health technologies. The “clinical” assessment is limited to
- the description of the health problem and the identification/description of current health technology,
- the examination of the technical characteristics of the health technology under assessment,
- the examination of its relative safety,
- the examination of its relative clinical effectiveness.
This means that other, non-clinical aspects – in particular the health economic issues and any added value related to pricing and reimbursement – will continue to be analysed and assessed by national bodies.
The HTA Regulation applies to medicinal products authorised using the centralised marketing authorisation procedure according to a staggered schedule (Article 7(1), letter (a), (b), Article 7(2)), starting with medicinal products with an oncological indication and advanced therapy medicinal products (2025). Orphan drugs will follow in 2028, then all other medicinal products in 2030.
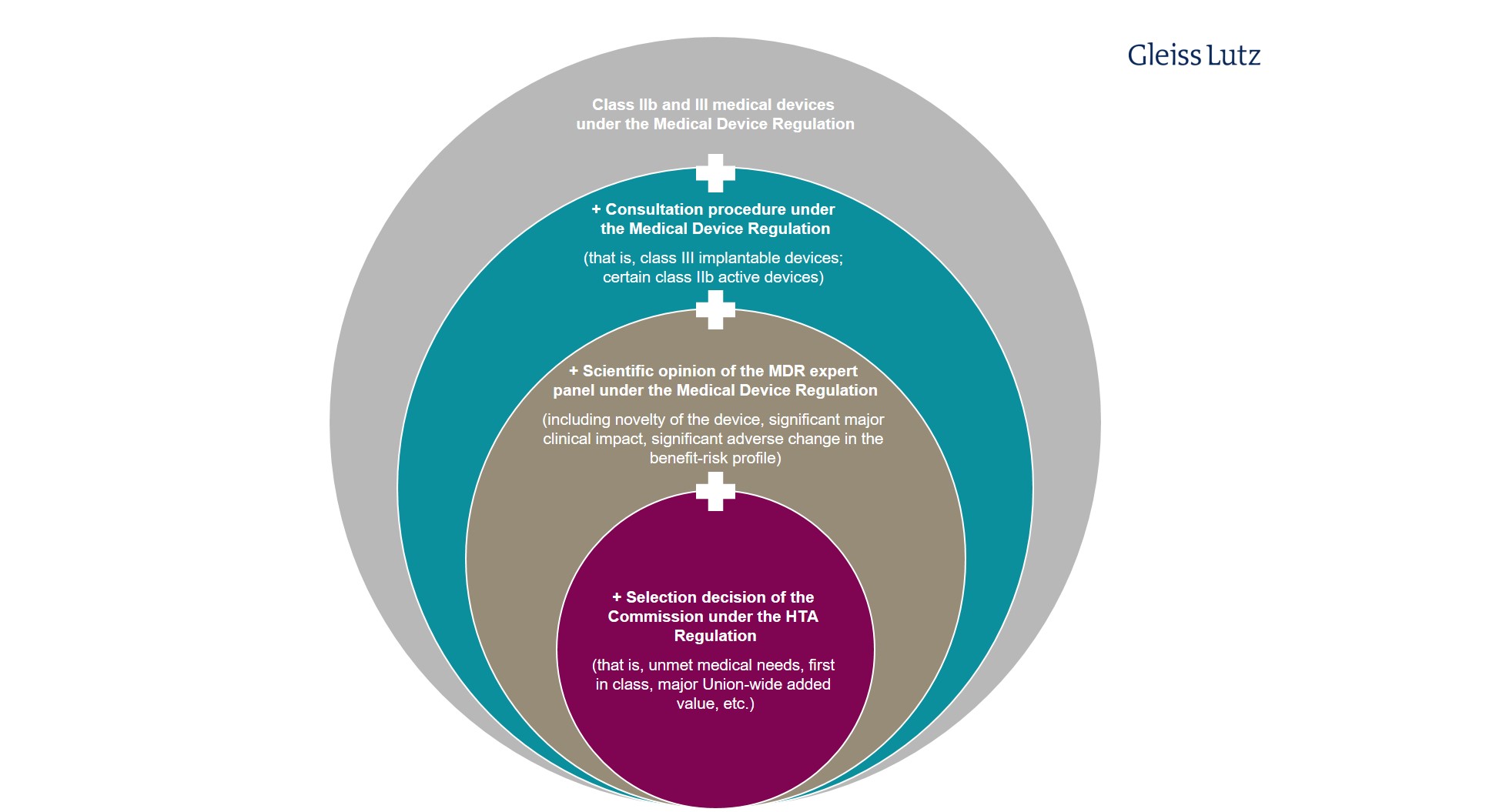
For medical devices and in vitro diagnostic medical devices, there are various filters restricting the HTA Regulation’s application (Article 7(1), letter (c)): Only particularly high-risk, novel, and/or likely high-impact implantable medical devices from class III and active devices from class IIb within the meaning of Regulation 2017/745 on Medical Devices (Medical Device Regulation, “MDR”) are clinically assessed at European level.
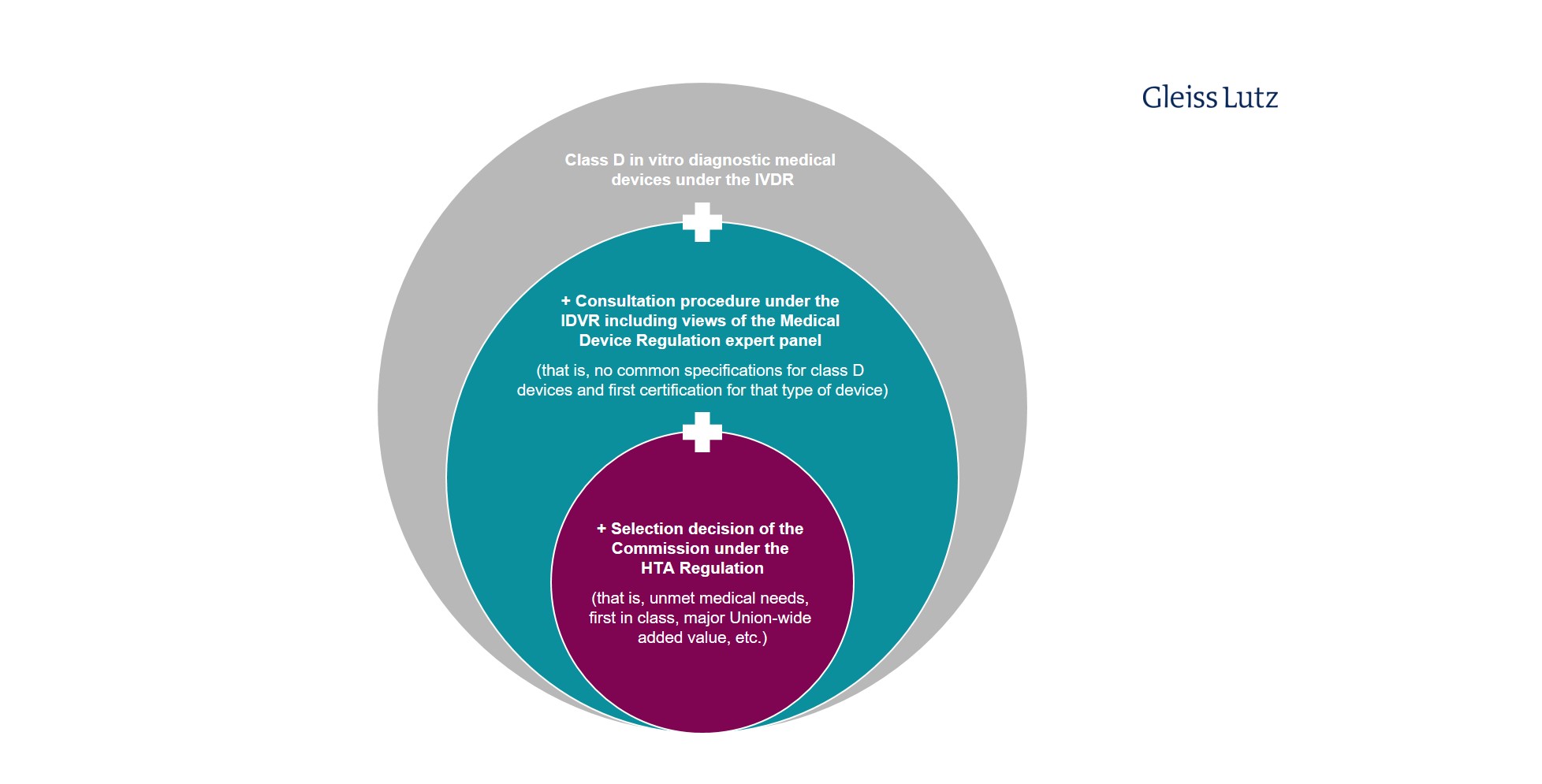
Similar restrictions apply to in vitro diagnostic medical devices. Only certain class D devices within the meaning of Regulation 2017/746 on In Vitro Diagnostic Medical Devices (IVDR) are covered by the HTA Regulation. Here you will find a graphic overview of the limited scope of application of the HTA procedure to specific in vitro diagnostic medical devices.
Overview of process steps
The joint clinical assessment is performed by a subgroup of the Coordination Group, which appoints an assessor and a co-assessor from different Member States from among its members. This is to ensure expertise and independence.
- The process starts with the definition of the assessment scope by the subgroup, taking into account the needs of the Member States and involving patients, clinical experts and other experts (Article 8(6)).
- The Commission then asks the developer to submit a dossier (Article 10(1)), the content of which – in the case of medical devices and in vitro diagnostic medical devices – is determined in more detail by Annex II of the Regulation and in Commission implementing acts. It is important that health technology developers do not submit evidence at national level that has already been submitted at Union level (Article 10(3)). Conversely, Member States may not request this evidence (Article 13(1), letter (d).
- The assessors then prepare drafts of a purely analytical assessment report and a summary report (JCA Report; Article 11(1)), which are limited to the relative effects of the health technology and the degree of certainty of the relative effects and may not contain any value judgement on the overall clinical added value (Article 9(1)). The assessment must take into account comments from patients, clinical experts and other experts, as well as from the other members of the subgroup (Article 11(3) and (4)). The company itself is only granted the right to report any purely technical or factual inaccuracies; comments on the results of the draft report are not allowed (Article 11(5)).
- The draft is then reviewed by the entire Coordination Group (Article 12(1)), endorsed by consensus, if possible (Article 12(2)) and, following procedural review by the Commission (Article 12(3)), published on an IT platform (Article 12(4)).
Timeline
Because the deadlines for the individual process steps in the assessments of medical devices and in vitro diagnostic medical devices are largely left to the Coordination Group, it is difficult to estimate the total duration of the joint clinical assessment. The Coordination Group’s procedural rules should contain more information on the timeframe.
Pain point: (Non-)binding nature of the assessment report becomes political
Unlike the Commission’s draft (COM (2018) 51 final), the HTA Regulation makes the clinical health technology assessment report non-binding. This issue was at the heart of the lengthy and contentious legislative process and prompted several Member States to raise subsidiarity complaints, including Germany. When carrying out national HTA, national bodies must now only give due consideration to the assessment report (Article 13(1), letter (a)). Moreover, the Regulation emphasises multiple times that the Member States’ competence to draw their own conclusions on the overall clinical added value and to make their own decisions on pricing and reimbursement on their own responsibility remains unaffected (Article 13(1), letter (a); recitals 14, 26, 28, 31).
Complimentary clinical analyses required for the general national HTA process and as non-clinical assessments by the Member States that are not covered by the HTA Regulation from the outset also remain permissible (cf. recital 15). Member States can also request additional evidence within this scope.
3. Joint scientific consultations (Articles 16-21)
For health technologies that are likely to be the subject of joint clinical assessments and where the clinical studies and investigations are still at the planning stage, health technology developers may request a joint scientific consultation with the Coordination Group. For example, the design of studies can be discussed with regard to patient populations, interventions, comparators and health outcomes (Article 16(1); cf. PICO process). Developers of medical devices may also request that the consultation take place in parallel with the process of obtaining scientific advice from the expert panels under Regulation 2017/745 on Medical Devices (MDR) (Articles 16(5) and 17(2)).
4. Voluntary cooperation (Article 23)
There will also be continuing support for voluntary cooperation between Member States on HTA and, thanks to the HTA Regulation, it will be embedded in a new organisational and financial framework. This most notably concerns the following:
- non-clinical assessments of health technologies under economic, legal, social, ethical and/or organisational aspects,
- clinical assessments on other medical devices and in vitro diagnostic medical devices that do not already come under the HTA Regulation in accordance with Article 7,
- HTAs for health technologies that are not medicinal products, medical devices or in vitro diagnostic medical devices (e.g. surgical procedures, disease prevention, or diagnosis and treatment).
5. Stakeholder network (Article 29)
The main opportunities for medical technology industry players to bring their influence to bear on European HTA exist through participation in what is known as the stakeholder network. This is being set up by the Commission from patient associations, consumer organisations, NGOs in the field of health, developers and health professionals. There is likely to be an open call for applications in the near future for this purpose. Applicants will be required to meet transparency obligations as regards sources of funding and other interests in the health technology development industry. The names of network members, and their transparency statements, will be made publicly available on an IT platform.
It remains to be seen how intense the exchange between the stakeholder network and the Coordination Group and its subgroups turns out to be in practice. The intention is simply for regular meetings to be held “at least once each year” (Article 29(5)). In addition, the Coordination Group will consult with the stakeholder network regarding the preparation of the annual work programme (Article 6(3), letter (d)) and as part of what is referred to as horizon scanning Article 22(2), letter (e)). Otherwise, the Coordination Group or its subgroups, as the case may be, will only consult with the stakeholder network upon request (Article 29(1)). The Coordination Group is also free to invite members of the stakeholder network to attend its meetings as observers (Article 29(6)).
III. Significance of the HTA Regulation for the reimbursement of medical devices under the German Social Security Code, Book V
The HTA Regulation assessments will have to be factored in at various stages of the decision-making processes on access and assumption of costs within the German healthcare system, most notably the following:
- Reimbursement for products classified as medical aids and the updating of the register of medical aids: This is based on the requirements set out in sections 33, 126 et seq. German Social Security Code, Book V (Sozialgesetzbuch, Fünftes Buch, “SGB V”) and directives on medical aids issued by the Federal Joint Committee. The register of medical aids (section 139 SGB V) lists medical aids for which, in the opinion of the National Association of Statutory Health Insurance Funds (GKV-Spitzenverband), reimbursement of costs is mandatory. One possible requirement for inclusion in the register may be evidence of medical benefit.
- New examination and treatment methods in panel medical and dental care (section 135(1), sentence 1 SGB V) and reimbursement based on a new German Uniform Evaluation Standard (Einheitlicher Bewertungsmaßstab, “EBM”) number: These methods are only reimbursable by the statutory health insurance funds if, upon application, the Federal Joint Committee has issued recommendations in the form of directives according to section 92(1), sentence 2, no. 5 SGB V, among other things regarding recognition of the diagnostic and therapeutic benefit of the new methods and their medical necessity and cost-effectiveness.
- The evaluation of hospital examination and treatment methods (section 137c(1) SGB V), which are verified by the Federal Joint Committee to determine whether they are necessary for the adequate, appropriate and cost-effective care of patients, taking into account the generally recognised state of the art as regards medical knowledge.
- New hospital examination and treatment methods using high-risk medical devices (section 137h(1) SGB V) and additional payment for them: Where first-time requests are made for additional payment under section 6(2), sentence 3 Hospital Fees Act (Krankenhausentgeltgesetz, “KHEntgG”) to the Reimbursement Institute (Institut für das Entgeltsystem im Krankenhaus, “InEK”) for a new examination or treatment method whose technical application is based largely on the use of a medical device that is classed as high-risk (known as NUB requests (NUB = “neue Untersuchungs- und Behandlungsmethode”, new examination and treatment method)), the hospital submitting the request, in agreement with the manufacturer of the medical device, must at the same time provide the G-BA with information on the state of scientific knowledge on this method for evaluation purposes.
IV. Outlook
The HTA Regulation is an important step towards a uniform assessment of innovative high-risk medical devices in the health insurance systems of the Member States, though it is ultimately up to the Member States to decide whether to assume the costs.
The fact that – contrary to the Commission draft – joint clinical assessments do not have binding character does not mean that the HTA Regulation will not create real added value because the national HTA bodies are directly involved in clinical assessment at European level via the Coordination Group. In practice, therefore, as far as the reimbursement procedures at Member State level are concerned, it will require considerable persuasion to call a positive EU-level HTA assessment into question at national level, perhaps even acting contrary to other Member States in the process. The Federal Joint Committee is already assumingthat the joint clinical assessment at European level must be factored into the early assessment decision on the basis of SGB V on added value for patients, even if this does not formally pre-empt the reimbursement decision.
From the point of view of the manufacturers of medical devices, the possibility of consultation – which ties in with the Medical Device Regulation – also represents an option to obtain, at an early stage of development, a preliminary assessment of products that are still in development, much in the same way as is already possible for pharmaceuticals.
Though this does not mean formally that the HTA assessment calls into question the sovereignty of the Member States, acceptance of the EU HTA among the Member States can be expected to grow as the HTA Regulation gradually gains traction.

{kind=link}
{kind=link}
{kind=link}